Undergrad Research Project: Antibiotic Resistance
Antibiotic resistance is a huge problem, especially in my country, Taiwan. Due to the health insurance policy, we have inexpensive access to medical care, resulting in overuse of antibiotics and emergence of resistant pathogens. During my rotation in the Intensive Care Unit, I saw a number patients fighting against resistant pathogens. Once patients have carbapenem-resistant pathogens, we are left with few choices. Health and technologies had advanced so much, but people are still dying from untreatable infections. Therefore, I chose to investigate resistance as my undergradate reseach.
advisor: Dr. Yu-Wei Wu at Graduate Institue of Biomedical Informatics, Taipei Medical University
A Pan-genome Based Network to Assess Resistant Potential of Hypothetical Genes
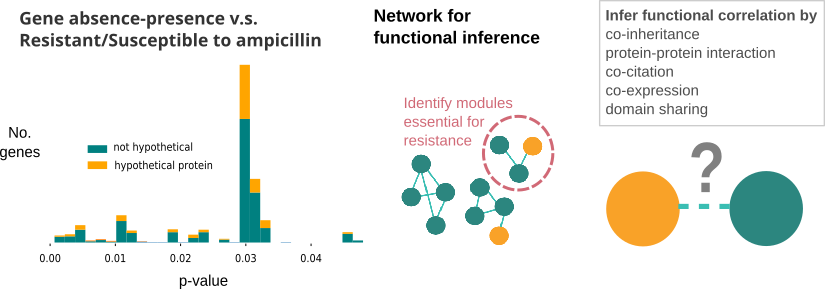
In our previous study, we used student’s t-test to identify resistance-related gene clusters within the E.coli pan-genome. Within the identified gene clusters, some of them were hypothetical proteins. We wanted to know about their functions, and how their presence/absense leads to resistance, so that new drugs can be made based on that.
Networks are very powerful methods to find out pathways or genes related to disease. There are co-expression networks, protein-interaction networks, and even integrated versions of them (termed co-functional network), such as EcoliNet, PseudomonasNet. However, when we queried the hypothetical genes using those built networks, it turned out those networks did not contain hypothetical proteins of interest.
To infer functional relation of those hypothetical genes, I proposed to rebuild the co-functional network, which we planned to include all the representing genes of the pan-genome. Co-functional network is based on six types of information: co-citation, co-expression, protein-protein interaction, protein domain sharing, co-inheritance and gene distance. Of all the six types of information for functional inference, co-inheritance has the best chance to provide functional linkage, as we could expect other kinds of data for hypothetical genes would be scarce.
To build co-inheritance network, a group of target genome has to be selected. The previous co-functional networks were based on reference genomes of prokaryotes, archaea, fungi and sometime eukaryotes. However, when searching for hypothetical protein homologs, those genomes generated no hits. We found out most homologs were harbored in closely related species, in this case are Enterobacteriae, Acinetobacter and Pseudomonas. Thus, ~2,000 genomes of those species were added as an additional set of target genome.
However, this additional set of target genome had very strong phylogenetic structures within, which would interfere co-inheritance analysis by confusing us with evolutionary events that occured at the root of the phylogenetic tree. We planned to integrate phylogenetic independent contrast to correct this. Other limitations of our study include: unable to address the difference within a gene cluster, and problems related to local statistical methods, as described in this review.
The importance of this work is to build a model for functional inference of hypothetical proteins, and assess the resistant potentials of them. The inferece made could guide experimental validation of the function. Future directions include: identification of novel resistance pathway, modelling different genetic aberrations (SNV, gene amplification, horizontal gene transfer).
Project: Predicting Antibiotic Resistance Based on Pan-genome
We aimed at building an integrated method of to predict antibiotic resistance based on interpretable feature and model. Previous prediction models were either based on uninterpretable features such as k-mer, de-burjin graph, or knowledge-based prediction.
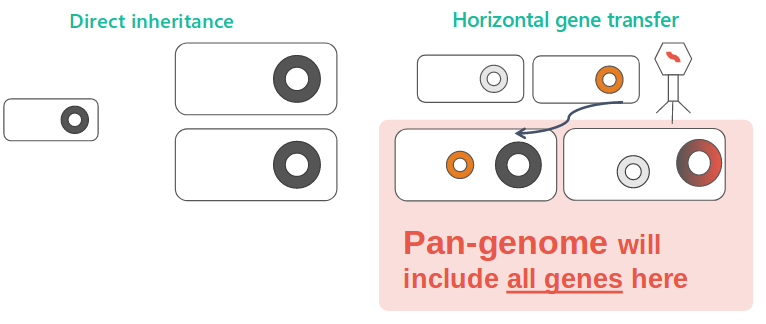
Given the importance of horizotal gene transfer, the pan-genome, a concept describing all possible genes by a species, is the excellent way to view resistance. We built a model based on the absence-presence pattern.
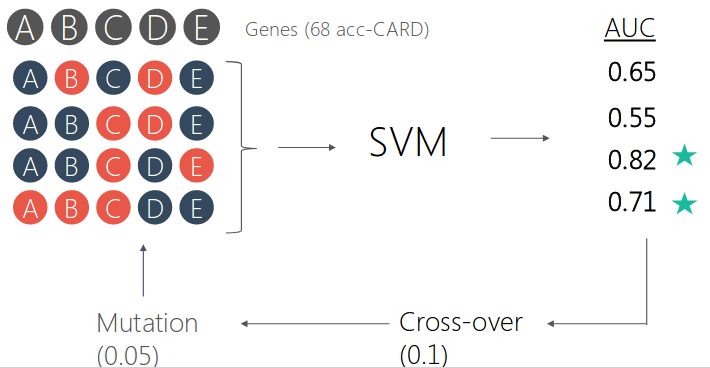
We select the best set of genes using genetic algorithm for the prediction. For each antibiotic, the model was built based on the selected set of features.
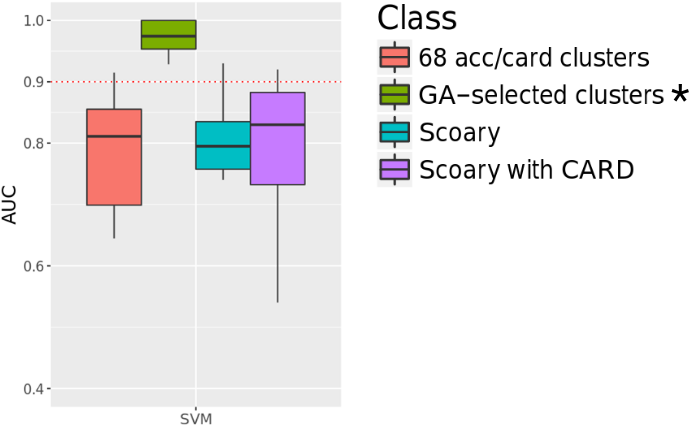
We antiticipate the selected features to be correlated with antibiotic mechanism of action. But it did not turned out the way we thought.
In this study, genomes with resistance annotation was downloaded from PATRIC during 2017. At the time of project initiation, the data was relatively scarce. But now there are more and more data, for better prediction and feature selection. Other limitations include: the gene pools for genetic algorithm selection was limited to known resistance genes; Single nucleotide variation and sequence difference within gene clusters were not considered; Potential information leakage during genetic algorithm interation;the gene selected from genetic algorithms were not explainable by current knowledge of drug mechanism.
We proposed a framework to select the best set of gene clusters for resistance prediction. I hope future models can become both interpretable and explainable, serving a guide for novel mechanism discovery.